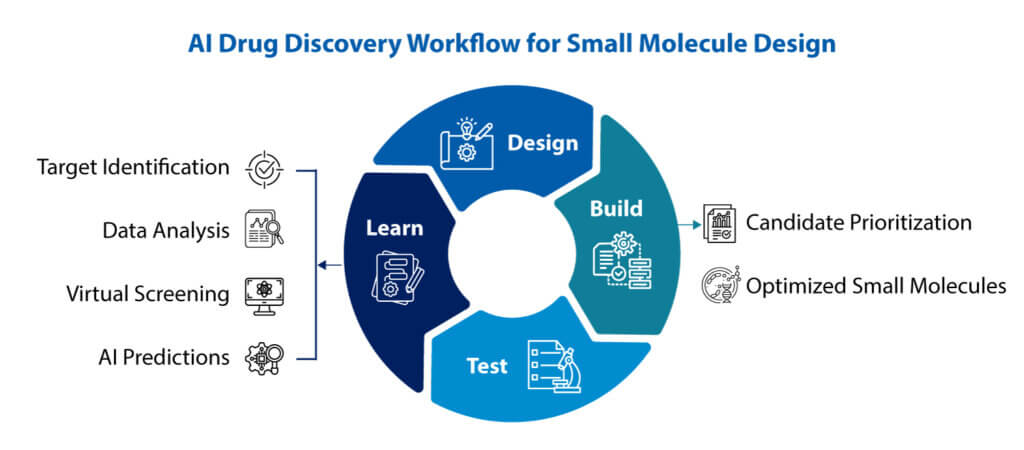
AI-driven drug discovery is changing how small-molecule programs are conceived, prioritized, and advanced. The older model relied heavily on sequential screening, manual hypothesis building, and long drug discovery cycles. That approach still has value, but it is no longer enough when target biology is complex, timelines are tighter, and failure is expensive. Today, AI drug discovery is pushing teams toward a more connected, data-led, and experimentally grounded way of working, where molecular ideas are generated faster, filtered earlier, and refined with better evidence.
Why small molecule design is being redefined
Small molecules remain central to modern therapeutics because they can enter cells, modulate a wide range of targets, and often offer practical development and manufacturing pathways. But designing a successful molecule has never been a simple chemistry exercise. Potency alone is not enough. The molecule must also show selectivity, acceptable physicochemical behaviour, workable ADME properties, manageable safety signals, and a route to synthesis that can scale. That is where AI drug discovery has become genuinely useful, not as a replacement for scientific judgment, but as a way to handle more variables at the same time.
Traditional discovery often moved in a fairly linear manner: identify a target, screen compounds, optimize hits, and hope the next round fixes what the last round missed. In practice, this created friction. A compound that looked strong in one assay could fail on permeability, metabolic stability, or synthetic tractability. AI and ML do not remove that uncertainty altogether, but they do help reduce iterative process. They can rank compounds, predict properties, flag structural liabilities, and detect patterns in large biological and chemical datasets that are easy to miss in conventional analysis.
Cheminformatics and multiparameter thinking in small molecule design
One of the clearest shifts has come through cheminformatics. For years, cheminformatics has supported library design, similarity searching, clustering, and structure-activity analysis. Now it sits much closer to the centre of decision-making. In AI drug discovery, cheminformatics is not just about organizing compounds. It is about learning from every synthesis, assay result, and failed series so that the next design round begins from a better position.
This matters because small molecule design is really a multiparameter optimization problem. A medicinal chemist may need to improve potency without worsening lipophilicity, reduce clearance without hurting solubility, or gain selectivity without making the scaffold too complex to synthesize. That is a more realistic view of discovery than chasing a single parameter. It reflects the fact that useful molecules are rarely the strongest in one property alone. They succeed because they balance several properties well enough to move forward.
Generative models, graph-based methods, and target-aware prediction tools have added another layer to this process. They can propose novel structures, prioritize analogs, and refine compound ideas much faster than manual enumeration. Still, novelty by itself is not the goal. The real value lies in producing a smaller, better-ranked set of compounds that medicinal chemistry and biology teams can evaluate in the lab. Some generated molecules may look elegant on screen and still fail when actual chemistry, biology, or scale-up constraints enter the picture. So, the output must remain tied to experimental reality.
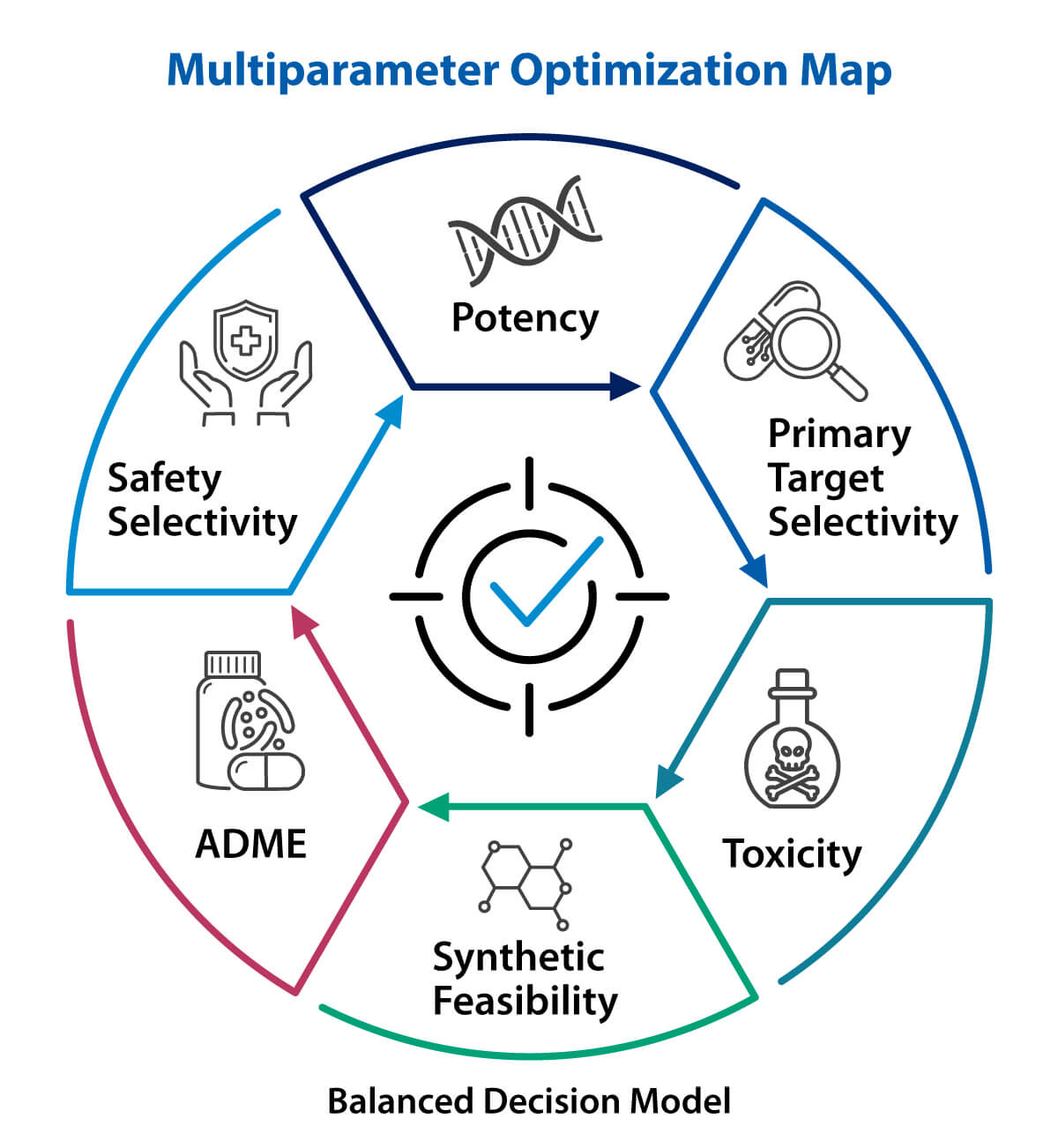
Figure 2. Cheminformatics strengthens small molecule design by balancing potency, ADME, safety, selectivity, and synthetic feasibility.
Design-build-test-learn and the rise of integrated drug discovery
The real paradigm shift is not only better prediction. It is the compression of the design-build-test-learn cycle. In a stronger integrated drug discovery model, data from target biology, screening, chemistry, DMPK, and early safety do not remain trapped in separate functions. It feeds a continuous loop. Molecules are designed with more context, built with fewer dead ends, tested with sharper hypotheses, and learned from in a way that improves the next cycle almost immediately.
This is where AI drug discovery begins to change operating models, not just software stacks. Teams can move from a handoff culture to a feedback culture. Computational chemistry, assay biology, medicinal chemistry, and data science stop acting like distant support functions. They become part of one decision system. A model is only as useful as the biological relevance of the data behind it, and the speed with which its predictions can be tested.
The same logic extends beyond virtual design. High-throughput screening, in silico ranking, and advanced automation are increasingly being used together, which expands the role of AI from target-related questions into lead generation, and broader early development decisions. So the shift is not from computation to chemistry. It is from fragmented discovery to integrated drug discovery.
What AI drug discovery still cannot do
It is easy to overstate the technology. AI models are only as reliable as the data they learn from. In drug discovery, that data is often noisy, biased toward successful series, generated across different assay conditions, or too sparse for confident generalization. A model may perform well on historical compounds and still struggle with a new chemotype or a difficult target class. Synthetic accessibility is another recurring problem. Some generated molecules may appear highly promising at the design stage and turn out to be impractical in the lab.
There is also an organizational issue. AI works best when data infrastructure, annotation quality, assay design, and scientist adoption are all mature enough. Without that, even strong algorithms produce patchy values. AI needs an operational backbone. Otherwise, it remains a pilot exercise rather than a true discovery engine.
Why AI in cell line development still matters here
At first glance, AI in cell line development may seem far removed from small-molecule design. However, in reality, it is not. The common lesson is that R&D value grows when data is made usable across steps, not when AI is treated as a narrow point solution. Whether the task is molecule design, target prioritization, process optimization, or cell line selection, the winning model is broadly the same: better data capture, stronger prediction, faster experimental confirmation, and disciplined iteration. That larger way of working will shape the future of integrated drug discovery across modalities.
Where the field is heading
The future of AI drug discovery in small molecules will likely be more hybrid than fully autonomous. Physics-based modeling, cheminformatics, generative AI, screening data, and medicinal chemistry judgment will work together, not separately. The organizations that gain the most will be those that can connect target biology, chemistry design, assay systems, and decision workflows in one practical framework.
In that sense, the future of small molecule design is not simply faster molecule generation. It is a better scientific selection, earlier visibility of risk, and a much tighter link between data and action. That may sound like a quiet shift, but it is a deep one. It changes how decisions are made, how teams work, and how quickly promising chemistry can move toward real development.
In conclusion, the AI Medicinal Chemistry Summit needs to firmly stand upon a tripod comprising a subject matter expert (medicinal chemist), a pertinent algorithm, and quality data. If any one aspect of the tripod falters, it will lead to the collapse of AI-led drug discovery efforts. Cognizant of this fact, Sygnene created a Centre of Excellence for holistic AI capability development.
Empowering Scientists at Syngene with a new Centre of excellence: AI Medicinal Chemistry Lab
Syngene Discovery Chemistry has created an AI Medicinal Chemistry Laboratory. This laboratory will include a dedicated server for the Discovery Chemistry team, relevant AI algorithms, and software backed by good-quality data. It will be operated by subject matter experts, including medicinal chemists and data scientists.
The platform will enable scientists to access and use key medicinal chemistry software for molecular design, visualization, docking, retrosynthesis, ADME prediction, and other tools for the efficient execution of the drug discovery cycle.
This initiative will also serve as a learning and development center for young medicinal chemists by providing hands-on learning experience, enhancing their computational capabilities, and improving their AI literacy.
For integrated drug discovery, this center will provide a strong foundation for training and knowledge sharing across chemists, biologists, and DMPK scientists. It will help accelerate design and decision-making in drug discovery projects and provide clients with a seamless drug discovery experience.