Oligonucleotide analytical development has become one of the more demanding parts of nucleic acid drug development. A short sequence may look simple on paper, but the analytical questions are not simple. Identity, purity, impurity identification and characterization, sequence integrity, backbone chemistry, modified sugars, conjugation, degradation, biological recovery, and method transfer, all need careful control.
The difficulty is not only technical, but also procedural. Many programs still treat oligonucleotides as if they can be managed with modified small-molecule analytical habits. That assumption is costly. A method may confirm the expected mass but miss a coeluting impurity. A batch may show acceptable purity, but closely related impurities may remain unidentified, poorly separated, or insufficiently controlled. A bioanalytical method may work well in a clean buffer system but lose accuracy in plasma, tissue, or other biological samples because of binding, degradation, extraction loss, or matrix interference.
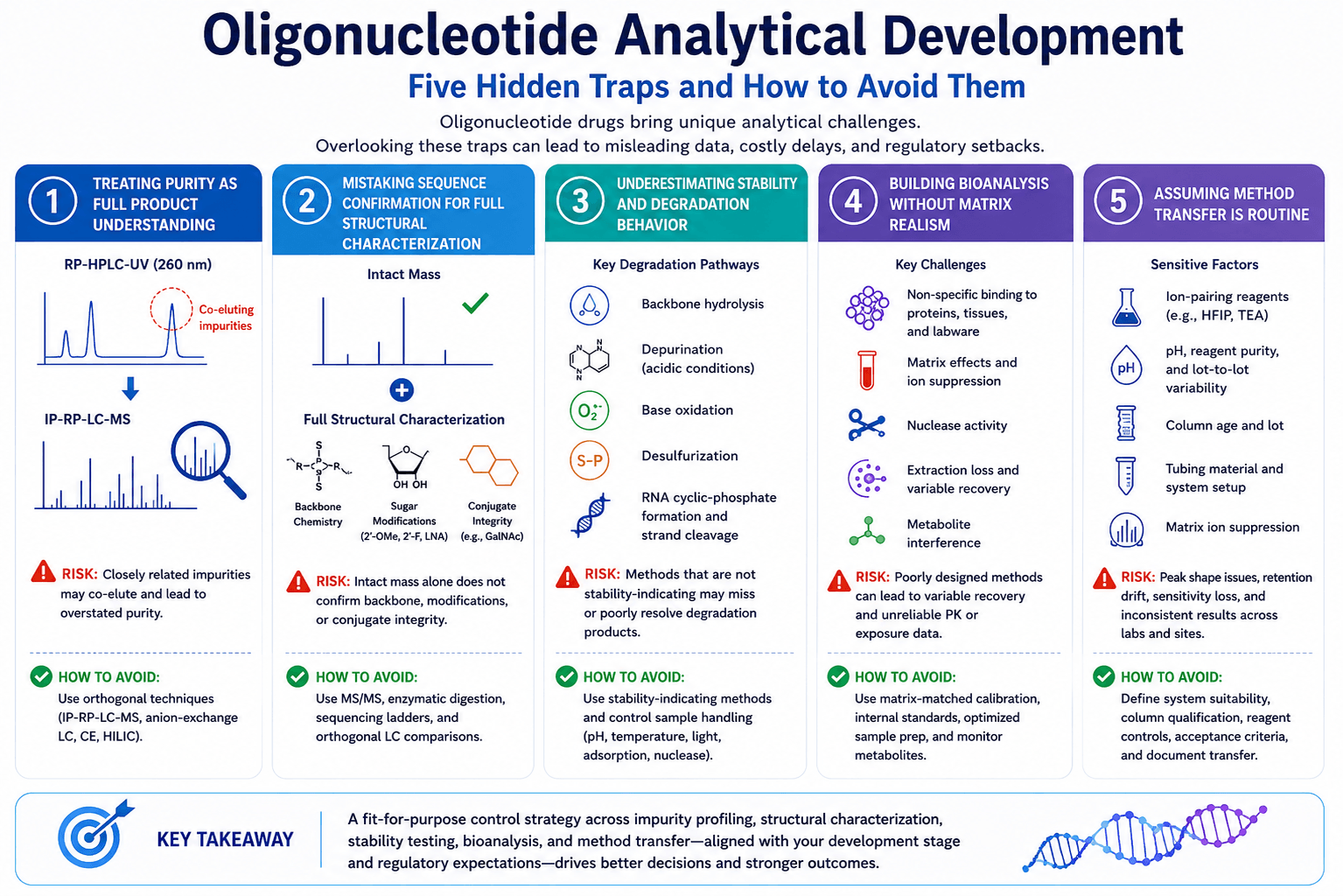
Figure 1. Five hidden traps in oligonucleotide analytical development, from purity assessment and structural characterization to bioanalysis and method transfer. (*AI generated image, for illustration purpose only)
For therapeutic oligonucleotide manufacturing, these are not small gaps. They can affect batch release, stability interpretation, toxicology material, clinical supply, and regulatory review. A strong development program therefore needs oligonucleotide synthesis, purification, characterization, stability testing, bioanalysis, and analytical method validation to be planned together at an early stage.
Why oligonucleotide analytical development needs its own discipline
Oligonucleotides do not behave like conventional small molecules, and they do not fit neatly into biologics workflows either. They are chemically synthesized, but their size, charge, sequence dependence, and biological handling create a separate analytical problem.
The impurity profile itself can be complex. Shortmers, longmers, depurinated species, deaminated products, oxidized products, desulfurized impurities, incomplete sulfurization products, protecting-group adducts, failed coupling products, and conjugation-related variants may all be relevant to characterize for a fully develop and robust method. Some separate clearly but some impurities might hide under the main peak. Some are visible by mass spectrometry but not by UV detection.
This is why a control strategy based only on one purity method is weak. A stronger approach combines oligonucleotide impurity profiling, LC-MS oligonucleotide analysis, stability-indicating methods, and phase-appropriate validation. The aim is not to generate more data but to know which data can support the next development decision.
Trap 1: Treating purity as full product understanding
Purity is an important starting point in analytical development, but it should not be read as complete product understanding. A reversed-phase HPLC-UV method at 260 nm may show a strong main peak and only a few smaller impurity peaks. On paper, that can look reassuring. The problem is that closely related oligonucleotide species may not always separate cleanly from the main product. When this happens, the main peak can carry more than one species, and the reported purity may look better than the actual product quality.
This risk is higher for phosphorothioate-modified oligonucleotides and other modified sequences. Incomplete sulfurization products, n-1 and n+1 sequences, depurinated species, deaminated products, and other near-neighbor impurities can behave very similarly during chromatography. A routine purity method may detect that something is present, but it may not show exactly what it is or whether it is fully separated from the intended full-length product.
. The answer is to use orthogonal methods that each address a defined risk. Ion-pair reversed-phase LC-MS (IP RP LC-MS) can support mass-based impurity identification. Anion-exchange chromatography can help separate charge-related variants. Capillary electrophoresis can add size and charge-based resolution. HILIC and high-resolution MS may provide further insight into selected molecules. Together, these methods can turn a simple purity result into a more reliable impurity profile.
This distinction is important. Purity answers one question: how clean does the product appear under a specific method? Product understanding answers a larger question: which related species are present, how well are they separated, and whether they matter for the process, specification, and regulatory package. Overall, a combination of techniques like IPRP-HPLC, supported by UV and LC-MS (Including full MS and MS/MS high-throughput analysis) identification is essential for identification and characterization of co-eluting or not well-resolved impurities.
Trap 2: Mistaking sequence confirmation for full structural characterization
Mass confirmation is often treated as a major milestone. It is useful, but it is not the same as full structural characterization. Intact mass may confirm that the expected molecule is present, but it may not prove that the backbone chemistry, modification pattern, or conjugate structure is fully correct.
This matters for antisense oligonucleotide characterization. A modified therapeutic oligonucleotide may require confirmation of phosphodiester and phosphorothioate linkage patterns, sugar modifications such as 2’-OMe, 2’-F, or locked nucleic acid units, and the integrity of GalNAc or other targeting conjugates. In conjugated oligonucleotides, the ligand or linker can also make interpretation harder because incomplete coupling or related variants may not be obvious from intact mass alone.
The approach assuming that one intact-mass result has answered every structural question is ultimately weak. A more reliable structural package may need high-resolution MS, MS/MS, enzymatic digestion, sequencing-ladder approaches, and orthogonal chromatographic comparison. The exact package will depend on the molecule and development stage.
Trap 3: Underestimating stability and degradation behavior
Oligonucleotide stability testing is not a simple copy of small-molecule stress testing. Oligonucleotides degrade through pathways that may include backbone hydrolysis, depurination under acidic conditions, base oxidation, desulfurization, strand cleavage, and, for RNA-containing constructs, cyclization-related degradation. The dominant degradation pathway is strongly sequence dependent.
The analytical method used for stability should be able to separate and measure relevant degradation products from the parent peak. If it cannot do this, the stability data may look orderly but still fail to support a regulatory specification.
Sample handling can also damage the result. Temperature, pH, freeze-thaw cycles, tube type, adsorption to plastic, residual salts, water content, light exposure, nuclease contamination, and storage time can all change the analytical picture. In a rushed program, these details are often overlooked and treated as operational housekeeping. That is a mistake. For oligonucleotide analytical development, sample handling is a fully integrated part of the method, not an afterthought. This is why, it is important to carry out forced degradation studies to understand the degradation pathway and characterize the impurities expected to emerge during the long-term storage and method ability to quantitate these impurities confidently.
Trap 4: Building oligonucleotide bioanalysis without matrix realism
Oligonucleotide bioanalysis should be designed around the biological matrix from the start. These molecules are charged, surface-active, and often measured at low concentrations. Phosphorothioate oligonucleotides can also bind non-specifically to plasma proteins, tissue components, and labware surfaces. If this binding is not controlled, recovery can vary from sample to sample. The data may then look biologically variable, when the real problem is the assay.
The practical solution is to build the method around the sample journey, not only around instrument response. Matrix selection, collection tube, anticoagulant, stabilization condition, storage time, freeze-thaw exposure, extraction chemistry, internal standard, and carryover control should be assessed early. A method that works in buffer should not be considered ready until it has been challenged in the actual matrix, such as plasma, liver, kidney, cerebrospinal fluid, tumor tissue, or cell lysate.
Method choice should also be linked to the question being asked. Hybridization-based assays can offer good sensitivity but may not always distinguish the parent oligonucleotide from closely related metabolites. LC-MS/MS can provide better structural selectivity but may need stronger sample cleanup and recovery control. In many programs, the best approach is not to choose a platform too early, but to compare what each platform can and cannot prove.
A more reliable bioanalytical plan defines the controls before the first pharmacokinetic or tissue-distribution study begins. These include recovery assessment, matrix effect evaluation, metabolite interference checks, carryover limits, stability during handling, and internal standards that track extraction performance. When these controls are built early, oligonucleotide bioanalysis becomes a decision-support tool rather than a source of late-stage doubt.
Trap 5: Assuming method transfer will be easy
Method transfer is one of the most underestimated risks in oligonucleotide analytical development. A method that performs well in one laboratory may behave differently in another, even when the equipment appears similar.
Ion-pair LC-MS methods are especially sensitive. Mobile-phase systems using hexafluoro isopropanol, triethylamine, or related alkylamine systems can be affected by pH, reagent purity, column age, source condition, tubing material, system cleanliness, and small differences in sample preparation. The result may be peak-shape changes, retention-time drift, carryover, loss of sensitivity, or matrix ion suppression.
This becomes more serious when the method moves from a development laboratory to a QC or GMP setting. The receiving lab may run more samples, use different reagent lots, follow different cleaning frequencies, or operate under tighter documentation requirements. Generic small-molecule transfer templates are often not enough.
A better transfer plan should include tighter system suitability criteria, column qualification, reagent control, sample-preparation details, acceptance ranges, troubleshooting rules, and clear documentation of what changed during transfer. Method transfer should be treated as a scientific activity, not clerical handover.
What these traps have in common
These five traps come from the same mistake: treating oligonucleotide analytical development as a small variation of familiar analytical practice when it isn’t. Oligonucleotides require dedicated thinking across impurity profiling, structural characterization, stability, bioanalysis, and method transfer.
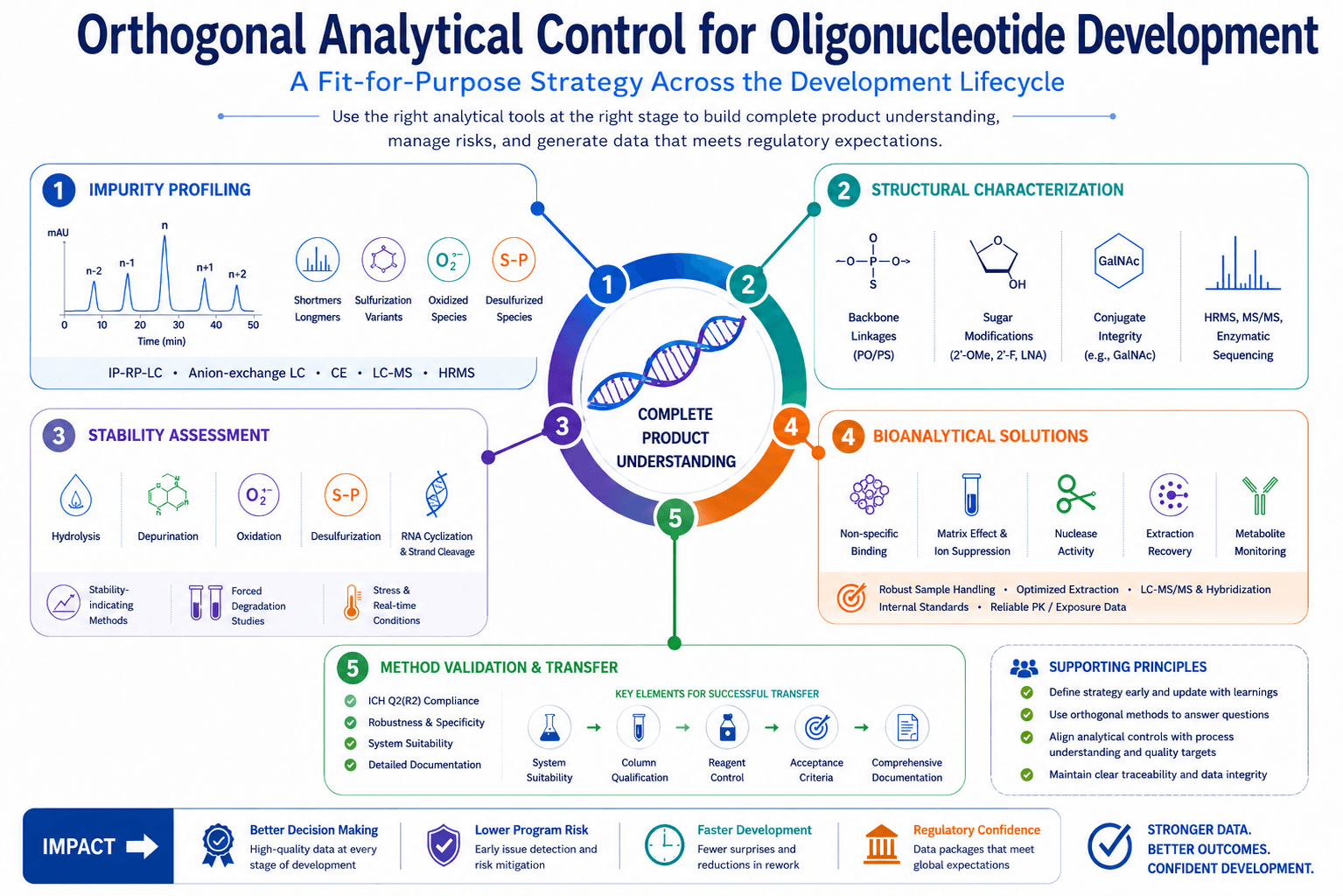
Figure 2. Orthogonal analytical control strategy for oligonucleotide development, covering impurity profiling, stability testing, bioanalysis, and method validation. (*AI generated image, for illustration purpose only)
The regulatory direction is also clear. Sponsors are expected to show that methods are fit for purpose at each development stage. Early discovery methods may be qualified lightly but methods used for release, stability, impurity control, toxicology-batch support, or clinical material need stronger validation logic. Specificity, precision, accuracy, recovery, robustness, carryover, sample stability, and matrix effect cannot be assumed.
For a CRDMO, the value is not only access to synthesis, purification, and analytical instruments. The real value lies in connected judgment. Therapeutic oligonucleotide manufacturing becomes more reliable when analytical development is linked with process understanding from the beginning.
Oligonucleotide analytical development works best when the team can explain what the method proves or not and when it must be strengthened. That is what prevents pleasant-looking early data from becoming uncomfortable questions later.