






Overview
Syngene, a leading global CRO/CDMO, partnered with a U.S.-based rare disease company to develop and manufacture a first-in-class treatment for a rare disease affecting children.
Syngene successfully developed the manufacturing process for the drug and supplied the required quantities to assist the company during different phases of drug development (preclinical and clinical) and later for commercial manufacturing.
The company received authorization for the sale of the product in the E.U. This was followed by regulatory approvals for sale in the U.S. and U.K. With this, the product has become the first drug in the world to be approved for children suffering from the rare disease.

The challenge
The company decided to partner with Syngene for process development and manufacturing of their product after developing it for several rare pediatric diseases. We received the discovery process for the synthesis of the product and set about developing the manufacturing process to improve the throughput and quality.
During this process, we encountered the following challenges:
- Need to redefine the regulatory starting material (RSM) strategy during the late stage of the development process
- Need for effective planning and execution to bring all crossfunctional teams onto a single platform for successful process validation
- Need for an in-depth technical understanding of the process and analytical methods to ensure flawless process validation
- Need for prompt troubleshooting support by all cross-functional teams to meet stringent project timelines
- Working during the Covid-19 pandemic with all its restrictions such as social distancing and masking, without affecting project timelines
- Need for a smooth transition to Syngene’s new digital systems (to ensure business-as-usual during a
pandemic) including effective, firsttime implementation as per project requirements
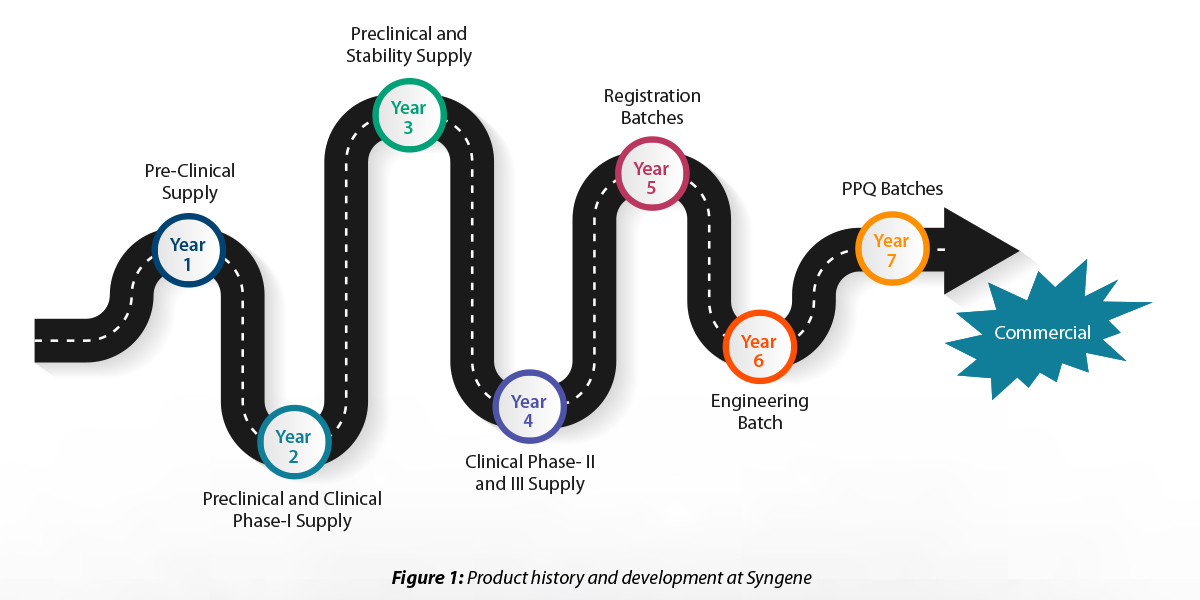
Product journey from Lab development to Drug Substance validation
Syngene played a pivotal role in the development of the product. The company required assistance in product development across different phases (preclinical and clinical). We manufactured and supplied the required quantity of the product in conjunction with the process development team.
Syngene’s approach
Defining strategies for designating RSMs
The product’s journey started with the development of appropriate regulatory starting materials (RSMs). We took up this challenge by undertaking process development studies on Syngene premises. This was followed by analytical method development and stability studies.
Later, based on the recommendations of the regulatory authorities, we came up with a strategy for designating the RSMs.
We quickly developed a process for the defined RSM and identified reliable vendors for the procurement of other RSMs. We then implemented this strategy for manufacturing registration batches of the product.
Process validation strategy
Based on our success with registration batches, we redefined the strategy for performing process validation (P.V.) or process performance qualification (PPQ) batches for the product.
A focused chemical development team was deployed to work on this program to enable the PPQ batches. During this period, the team successfully completed the pre-validation studies for the process and analytical methods. Subsequently, we completed the key phase of the program – engineering as well as process validation campaign for the product.
P.V. or PPQ validation is required to assure stakeholders that the processes will produce consistent and repeatable results within the predetermined specifications.
P.V. also verifies whether the quality standards and compliance requirements are being met by the product in realtime – an important consideration for any pharmaceutical facility.
To accomplish the PPQ campaign, we deployed a dedicated chemical development team from various crossfunctional areas. The campaign enabled the gathering of complete and in-depth information about control strategies, critical process parameters (CPPs), critical quality attributes (CQAs), scale-up parameters, and so on. We also undertook detailed pre-validation studies and assessment of existing campaign knowledge to ensure the success of the campaign.
Performing pre-validation studies
A substantial commitment is required to perform pre-validation studies, both in terms of time and resources. Another major requirement is to work in line with ICH guidelines to provide a defined and robust quality control strategy, including identifying CPPs and unit operations that
impact defined CQAs.
Activities/Studies | Outcomes |
|---|---|
Existing process and knowledge assessment | Process development summary report |
Process and analytical method development | Process validation and analytical method validation |
Identification and characterization of impurities | Synthesis of impurity reference standards and markers |
Spike purge/process control justification (PCJ) studies | Control strategies and critical quality attributes (CQAs) |
Process characterization, including design of experiment (DoE) | Robustness, critical process parameters (CPPs), normal operating
range (NORs), proven acceptable range (PARs) |
Analytical method (AM) verification including precision,
accuracy, limit of quantification (LOQ), limit of detection (LOD) | Analytical method validation, mechanism of action |
Process engineering and process safety studies | Equipment qualification, safety risk assessment |
Figure 2: Pre-validation studies – Overview
Experimental plan for process control
justification (PCJ)/Spiking studies | Defining experimental plans for spiking of impurities/intermediates |
Experimental execution for
PCJ/Spiking studies | • Spiking of impurities/intermediates and execution of the process • Identification of purge points in the process through analysis of isolates and waste streams |
Data summary and reiteration | • Compilation of data and identification of critical quality attributes • Reiteration of experiments to justify the limits (if any) |
Detailed PCJ/Spiking report | • Complete report, defining all studies, results, and conclusion on purging impurities |
Control strategy | • Defining specifications for raw materials, in-process controls, and intermediates, based on
studies |
Figure 3: Process control justification / Spiking studies – Overview to define the CQAs
Assessment of cause and effect (C&E)
matrix and failure modes and effect
analysis (FMEA) | • Study of current process data with respect to the impact on product quality by C&E and
FMEA |
Experimental plan/protocol for
process characterization studies (PCS) | • Further experimental plan/protocols developed with respect to NOR/PAR, DoE, OFAT, etc.
as appropriate |
Experimental execution of PCS plan | • Execution of experiments as per pre-defined protocols/plans at an appropriate scale and
analytical data to be generated |
Data summary and reiteration | Compilation of data and identification of critical process parameters (CPPs) NOR, PAR, etc • Reiteration of experiments to justify the process boundaries (if any) |
PCS Report | Complete report defining all studies, results, and conclusion with defined process
boundaries and CPPs |
Figure 4: Process characterization studies – Overview to define the CPPs
At Syngene, process validation also involves the collection and evaluation of data from the process development stage through to commercial production. This is done to establish the scientific evidence that a process is capable of consistently delivering quality products, including assurance of the reliability of supply. Based on successful pre-validation studies, we defined the process validation master plan and initiated the process validation campaign of the product.
Integrated approach to process validation
We implemented an integrated approach to execute the process validation campaign that required expertise across various disciplines. This included the process research and development (PRD) team / analytical research and development (ARD) team, the scale-up team comprising technology transfer, process engineering, production team, and the quality management team (quality control and quality assurance).
To ensure the success of the program, we prepared a detailed plan involving effective coordination with cross-functional teams and support from senior management. We successfully completed PV/PPQ batches along with pre-validation data with the defined CPPs and CQAs. This helped the company submit the final NDA documents to USFDA and EMA regulatory authorities.
NDA submission to regulatory authorities
The next step was to obtain the pre approval inspection (PAI) for the drug substance. Syngene played a significant role in helping the company complete this process, successfully. Following the PAI, the company submitted a marketing authorization application (MAA) to EMA and a new drug application (NDA) to the U.S. FDA seeking approval for the product. The European Medicines Agency (EMA) granted the product, accelerated assessment, orphan designation, as well as access to the PRIority MEdicines (PRIME) scheme. Similarly, the program received fast-track, rare pediatric disease and orphan drug designations in the United States. The product has since been approved in the U.S., E.U., and other countries.
Conclusion
Syngene’s association with the biotech company only goes to show how outsourcing critical services to a reliable partner can help accelerate drug development programs.
We not only helped to define RSMs for the product but also successfully developed the manufacturing process to improve the throughput and quality of the product.
Further, in conjunction with process development, we manufactured and supplied the required quantity of the product for preclinical and clinical supply, and more recently, for commercial manufacturing. We also provided support for submitting marketing applications for the product in various regions.
As a result of our combined efforts, the product has become the first drug in the world to be approved for children suffering from the rare disease.
To know more about our Drug Development services or to contact our experts, please click here.