






Disease overview
Cholestatic liver disease is a condition that causes liver damage and fibrosis due to bile stasis. It is characterized by the slowing or stalling of bile flow through the hepato-biliary system (liver and bile duct). When bile flow is affected, it backs up into the organs and bloodstream, caus ing inflammation. The disease is represented by primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC), but the pathophysiological pathways that cause bile stasis in both diseases are different. The pathogenesis of the disease is still unclear, although autoimmune mechanisms have been postulated and partially elucidated.
Although the disease may progress slowly with only mild liver dysfunction, symptoms of illness, jaundice, and severe itching, chronic inflammation of any of the organs can lead to permanent scarring, causing impairment of their function. If left untreated, cholestasis will lead to fibrosis, biliary cirrhosis, and ultimately, end-stage liver disease, which requires liver transplantation. As a medical treatment, ursodeoxycholic acid is widely used for PBC and has proved highly effective against disease progression. However, its efficacy is limited in cases of PSC, and research and development of various drugs for increased efficacy are still underway.
Non-alcoholic steatohepatitis (NASH) is another severe condition characterized by steato sis (build-up of fat in hepatocytes), concomitant liver inflammation (both are treatable), and fibrosis (which is untreatable). It is also caused by excess build-up of bile acid in the liver. It is part of a group of conditions called non-alcoholic fatty liver disease (NAFLD). NASH results in liver failure and cirrhosis. There is an unmet medical need for both chronic diseases (NASH and cholestasis), as no drug is available to treat these conditions

The requirement
A mid-size biotech company partnered with Syngene to identify a clinical candidate for treating cholestatic diseases. Syngene was tasked to develop a molecule to remove the bile acid from the ileum and liver without causing diarrhea.
The goal of the collaboration was to identify a development candidate for cholestatic disease with a novel mechanism of action that would stop the reabsorption of bile acid in the intestine and liver. Accelerated bile acid elimination reduces the bile acid concentration in the liver and systemic circulation.
The challange
- To design a compound that shows significant inhibition of ASBT and NTCP targets without causing diarrhea
- To optimize the structure-activity relationship of two of the client’s existing drugs to ensure optimal IC50 values and high bioavailability
Mechanism of action of the target
There are two cellular protein pumps present in the body – the ileum (intestine) and the liver, known as the apical sodium-dependent bile acid transporter and sodium/taurocholate co-transporting polypeptide, respectively. These pumps are transmembrane proteins that can inhibit cholestatic disease. Pharmacological ASBT inhibition results in decreased bile acid absorption in the intestine and, subsequently, higher bile acid elimination through faeces. This, in turn, results in reduced systemic and liver bile acid concentration and is expected to improve the cholestatic liver disease condition in animal models.
In humans, the client had tested small molecule ASBT inhibitors in clinical trials with varying indications. However, one of the side effects of this drug is that it causes diarrhea in some patients. This is currently being evaluated in BOLD global phase-3 clinical trials in patients.
Earlier, the client had also manufactured another drug for chronic constipation in cancer survivors and bedridden patients. The drug was found to increase bile acid in the ileum and relieve patients of reduced bowel movement. However, in other patients, this drug resulted in severe diarrhea.
For the development of the current molecule for cholestatic disease, Syngene was required to modulate the inhibitor activity of both the existing drugs (for treating cholestatic disease and chronic constipation) to help remove bile acid from the ileum and liver without causing diarrhea.
Target rationale
The target protein’s inhibition affects the liver’s bile acid metabolism with modulation of the bile acid levels in the serum and target organ. This
was evident from the preclinical findings and human population studies.
Medicinal chemistry: Path toward candidates
Syngene’s medicinal chemistry team adopted a ligand-based design approach to identify the lead compound through rational design, including exploring its detailed structure-activity relationship. The team committed to invest considerable time understanding the mechanism of action to improve the ADME profile of the lead analogues. The overall program demonstrated the high-level efficiency of the iterative designmake-test-analyze (DMTA) cycle to identify and optimize the lead analogues. The team successfully identified a lead optimized compound with low nanomolar IC50 and excellent in vitro pharmacokinetic (PK) profile with improved bioavailability.
The team successfully established the tractable SAR database by exploring various Med-Chem strategies:
- Variation in chain length which impacts potency and reduces lipophilicity thus PPB.
- Bio-isosteric replacement with acceptable potency, and evaluation of impact by changing the position.
- Impact of chirality on selectivity.
Syngene team selected lead compounds with single digit nanomolar potency and successfully optimized the compounds’ in vitro pharmacokinetic profiles.
The screening cascade used for the selection process is depicted below:
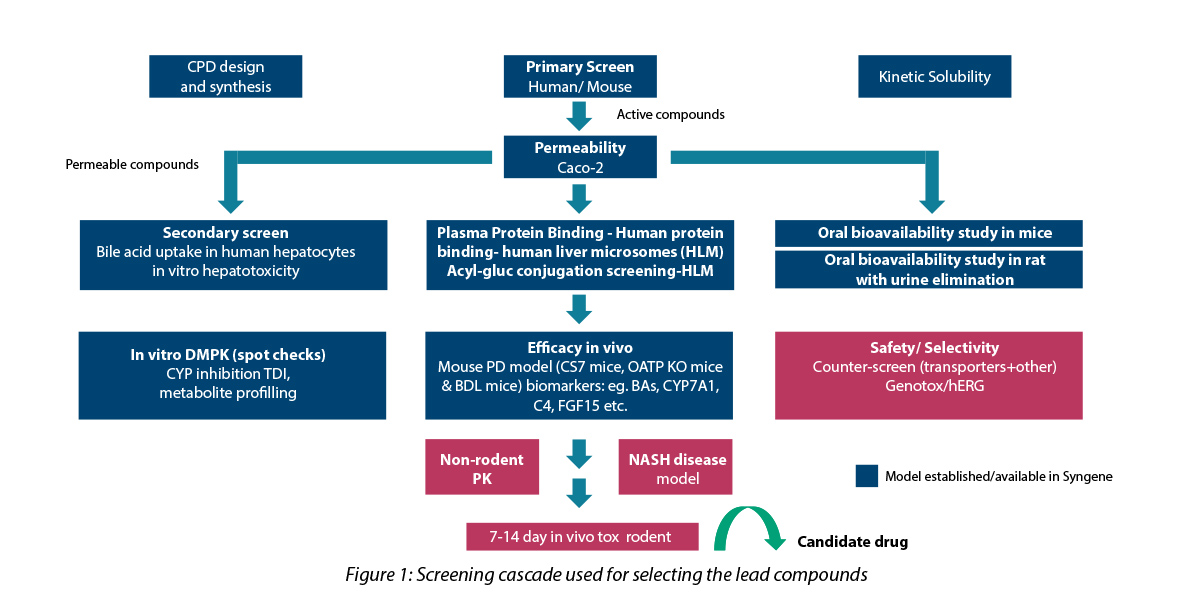
Key strategies employed in DMPK
- Rapid ADME profiling: To facilitate rapid screening, only kinetic solubility, plasma protein-binding and permeability assessment were included in the screening cascade for Tire-1 assays with rapid turn-around time.
- Soft spot analysis, and inputs to the synthetic chemist helped in metabolic site re-programming: The small molecule consisted of long-chain fatty acids. In the body, these hydroxylates are metabolized by specific enzymes and subsequently eliminated. By using metabolic site reprogramming, the drug molecule was structurally modified to reduce the rate of metabolism and thus increase exposure, thereby enabling the desired physiological effect. The structure-based design studies illuminated two sites for hydroxylation, which matched the predicted outcome (in silico and experimental data).
- Glucuronide conjugation: A terminal carboxylic acid functional group is necessary to retain activity. Hence, acyl glucuronide conjugate formation tendency was ruled out for in-vitro human liver microsome studies.
- Customization: Tailoring of study protocols was carried out for accurate rank ordering of test compounds. This facilitated early identification of lead molecule with desirable ADME properties.
Primary and secondary cellular (hepatocyte) screening for identification of hits and leads
- Cell-based assays were used to model the liver disease. The primary assay tested the binding capacity of the lead molecule to the target, and the secondary assay examined the inhibition of bile acid production. Assays were developed and validated using in-house generated cell lines.
- Cellular uptake assays using overexpressing cell lines were used to screen different compounds through IC50 comparison.
- A hepatocyte cell-based functional assay was used to evaluate the deposition of fats, which is one of the hallmarks of cholestatic disease.
- Percentage inhibition was compared in cellular systems — OE cells, HepaRG vs. primary human hepatocytes (PHH). HepaRG, is an immortalized hepatic cell line. High P450 activity and PHH are the gold standards for hepatic in vitro culture models. These were used for functional testing and for the selection of the compounds with efficacy in vitro NAFLD model.
- Cell images showed induced or reduced lipid accumulation in HepaRG cell line after being treated with the drug. Regular screening was carried out in a cell-based radioactive uptake assay for the primary target, selectivity assays, binding ability to both the targets and species ortholog assays.
PD study to measure biomarkers
For the identified lead compounds, PK & PD studies were done in C57BL/6 & OATP knockout mice. The compounds were tested for the following:
- Biomarker concentration at time points
- Serum bile acid elevation and increased fecal bile acid elimination.The lead compounds engaged with the target showed a dose-dependent increase in serum bile acid levels and increased elimination of fecal bile acid.
Gene expression levels of CYP7A1 (cholesterol biosynthesis feedback) and FGF15 (growth factor for lipid deposition) were used as the biomarker to study the effect of these inhibitors. It was found that the compound induced changes in the expression of these biomarkers and could alleviate symptoms of cholestatic disease and NASH.
Summary
Syngene successfully identified a first-in-class, potent and efficacious inhibitor for apical sodium dependent bile acid transporter and sodium/taurocholate co-transporting polypeptide targets for the potential treatment of cholestatic disease.
To achieve this, Syngene’s medicinal chemistry team designed several lead analogs and conducted studies to understand the structure-activity relationship. Lead molecules were identified by screening the analogues through various cell-based activity, ADME profiling, and PK-PD studies measuring various biomarkers in animal models.
The novel inhibitor drug has now progressed to the preclinical stage of development. The pharma company is looking forward to receiving regulatory approval soon.
To know more about our small molecule discovery solutions, contact our experts.