






Introduction
Transformative innovation in the pharmaceutical industry has resulted in an explosion in the types of novel modalities and therapies. The world of drug discovery and development is rife with new approaches within immunotherapy, personalized vaccines, microbiome therapies, and CRISPR-based therapies to provide novel treatments and cures for a wide range of diseases. Even though progress is being made in these areas, more than 90 % of the drugs currently available on the worldwide therapeutics market are small molecules based.
As recently as in 2017, 9 of the top 10 sellingmedicines in the US were small molecules, demonstrating why they are so important, even in our current-day personalized approaches to medicine.
The challenges involved in small molecule drug development are highlighted by the fact that only 1 out of every 10,000 compounds synthesized will eventually obtain FDA approval. The right strategy for early phase development can go a long way towards maximizing the chances of success for small molecule new chemical entities (NCEs).
An important lever in early phase drug development is Chemistry, Manufacturing and Controls (CMC) – all the activities that go into ensuring that a drug product is safe, effective and consistent. Designing and implementing a scientifically-sound, phase-appropriate CMC development strategy can reduce costs while building asset value. What we at Syngene propose is an integrated CMC strategy that will help achieve faster regulatory filing and Phase 1 First-in-Human (FIH) studies while helping maintain tight control over quality and spends.

An Integrated CMC strategy
Deep scientific expertise, a pragmatic approach towards achieving development objectives and timelines, and a comprehensive understanding of regulatory requirements must come together seamlessly in order to shape the CMC strategy. Rational pharmaceutical development with the integration of various drug development disciplines is the logical first step in this CMC journey. A key principle is that phase appropriate
regulatory and quality guidelines, design of pre-formulation studies, analytical methods, dosage form selection and development for toxicology and FIH studies should be considered in parallel to drug substance development. Given the complexity of CMC, the best way to develop a clear strategy is to base it on a holistic target product profile. An understanding, not only of the basic science of CMC, but how that fits into preclinical strategy, clinical strategy and regulatory strategy, is essential. This will ensure that a one-size-fits-all approach to CMC is not adopted. Anticipating potential problems and generating the right data sets to address those problems is the cornerstone of the integrated CMC strategy. Based on the above principles, Syngene has developed an integrated CMC approach which has been defined below:
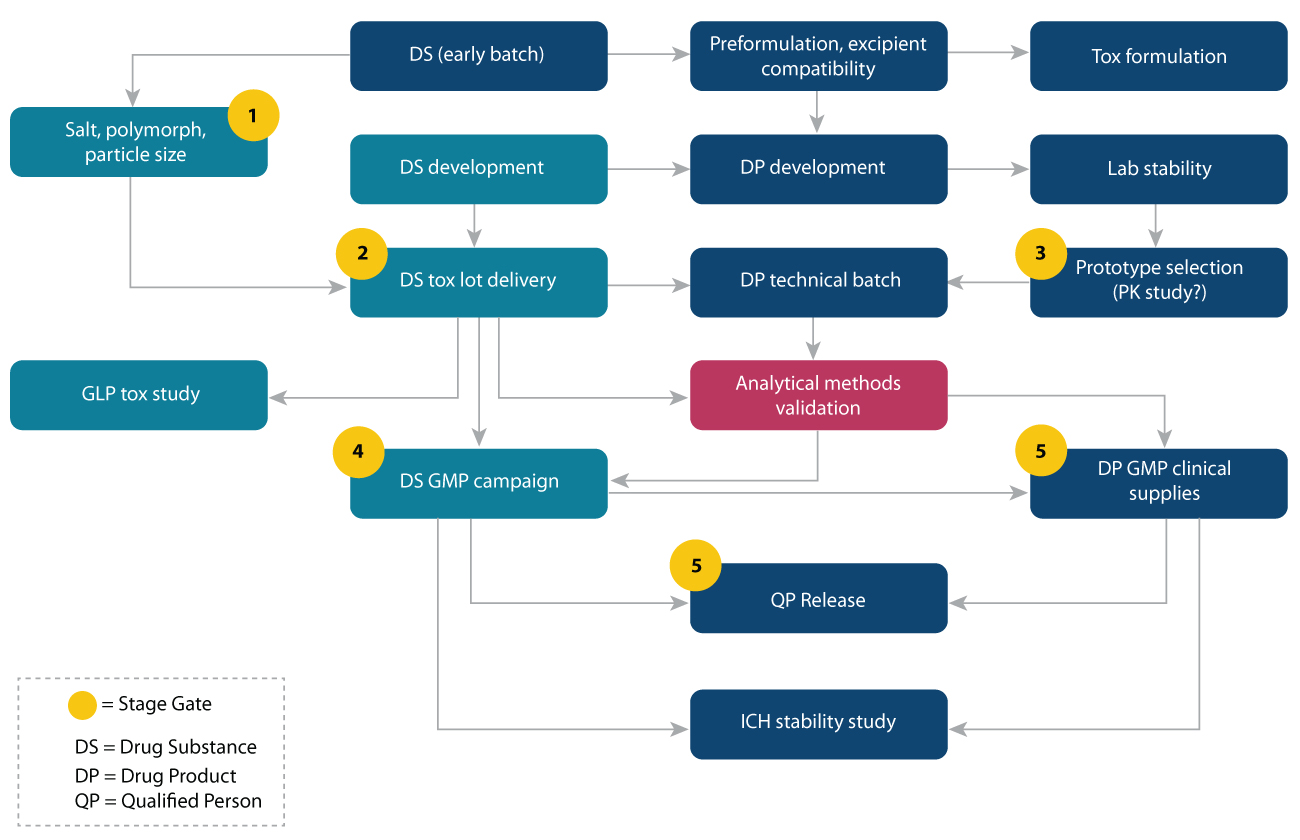
At Syngene, we have broadly classified the CMC strategy into 7 phases:
1. Regulatory strategy (including filing date)
2. Developability assessment
3. Drug Substance development strategy
4. Pharmacokinetic and GLP toxicology studies
5. Drug Product development strategy
6. Analytical Development & validation
7. Quality requirements
What does the integrated CMC strategy translate to?
Regulatory strategy + Quality requirements
The regulatory and filing strategy, as well as the quality requirements, should be considered at the beginning of development to avoid delay in approval from authorities for FIH study. To achieve this, an integrated cross-functional CMC development team with an understanding of quality and regulatory requirements is essential. This approach has been seen to enable rational, faster, and successful drug development of clinical candidates.
Developability assessment
The intrinsic physicochemical and biopharmaceutical properties of the molecule are vital considerations in designing a rational drug development program. A thorough understanding of these properties will help anticipate challenges in development which could potentially cost time and money down the line. Solubility and bioavailability evaluations are important parameters to be studied prior to nominating for clinical development. Screening and selection of salt form and polymorph are additional considerations. Other biopharmaceutical properties such as oral dose proportionality, Pgp efflux, clearance, Cmax, regional absorption, and food effect guide the dosage form development for toxicology and FIH studies.
Pharmacokinetic and GLP toxicology studies
Studying the fate of the molecule in the body and its potential harmful effects is another important phase in the CMC strategy. PK & Dose range-finding study, GLP 28-day repeat toxicity, QA-approved tox package, rodent & non-rodent tests, formulation and exposure (including bioavailability and dose limitations) provide insights which determine the suitability of the molecule for further development.
Drug Substance development strategy
The drug substance development phase is possibly the most critical aspect of drug development. The quality, quantity, and timely availability of the drug substance are critical considerations during development. The route of synthesis selected should avoid hazardous reagents, toxic impurities, toxic solvents, and low yield, and yet produce compounds with desired quality attributes such as the right particle size distribution and impurity profile. Identifying correct regulatory starting materials (GMP steps) and appropriate quality specifications at each step is essential, as is a concrete risk assessment.
Drug Product development strategy
The physicochemical, pharmacokinetic, and clinical study design considerations are the parameters that need to be strongly considered for drug product selection, design, and development. The simple and conventional formulation options such as drug-in-capsule, drug-in-bottle for onsite compounding, and immediate-release capsule/tablet should be given preference, provided the compound shows good solubility and dose proportionality for oral dosing. However, the need of an enabling formulation should be identified early on through animal PK studies to achieve adequate exposure for toxicology and FIH studies, as in both the cases several doses are administered in an escalated manner to characterize NOEL (No Observed Effect Level) and MTD (Maximum tolerable dose). The clinical drug product specifications should be generated with due consideration of the specifications of the toxicology batch of the drug substance, especially with respect to impurities.
Analytical Development
The drug substance and drug product methods should be developed in tandem with an emphasis on the impurities method – the objective is to develop one common method for both to minimize impurity isolation and characterization. Preliminary and limited forced degradation studies can be developed as stability indicating methods. Other methods specific to drug substance (residual solvents, solid-state, particle size, heavy metals, counter ion content) and drug product (dissolution) need to be developed as well. A phase-appropriate approach to verification/validation is justifiable before regulatory authorities during early phase development. The stability data generated using developmental lots of drug substance and drug product can be used for filing while the formal stability study is being conducted as per ICH, which can be amended later. This approach helps in the faster regulatory filing of IND/IMPD.
The integrated CMC strategy enables expedited IND/IMPD filings and FIH studies, with typical time to regulatory filing being ~11 months. This is primarily because the approach, when coupled with deep scientific expertise that a CRO like Syngene has, makes early identification of challenges possible. This allows for adjustments to be made without asset value loss and enables risk mitigation through the end to end consideration of regulatory strategy before the start of the development program.
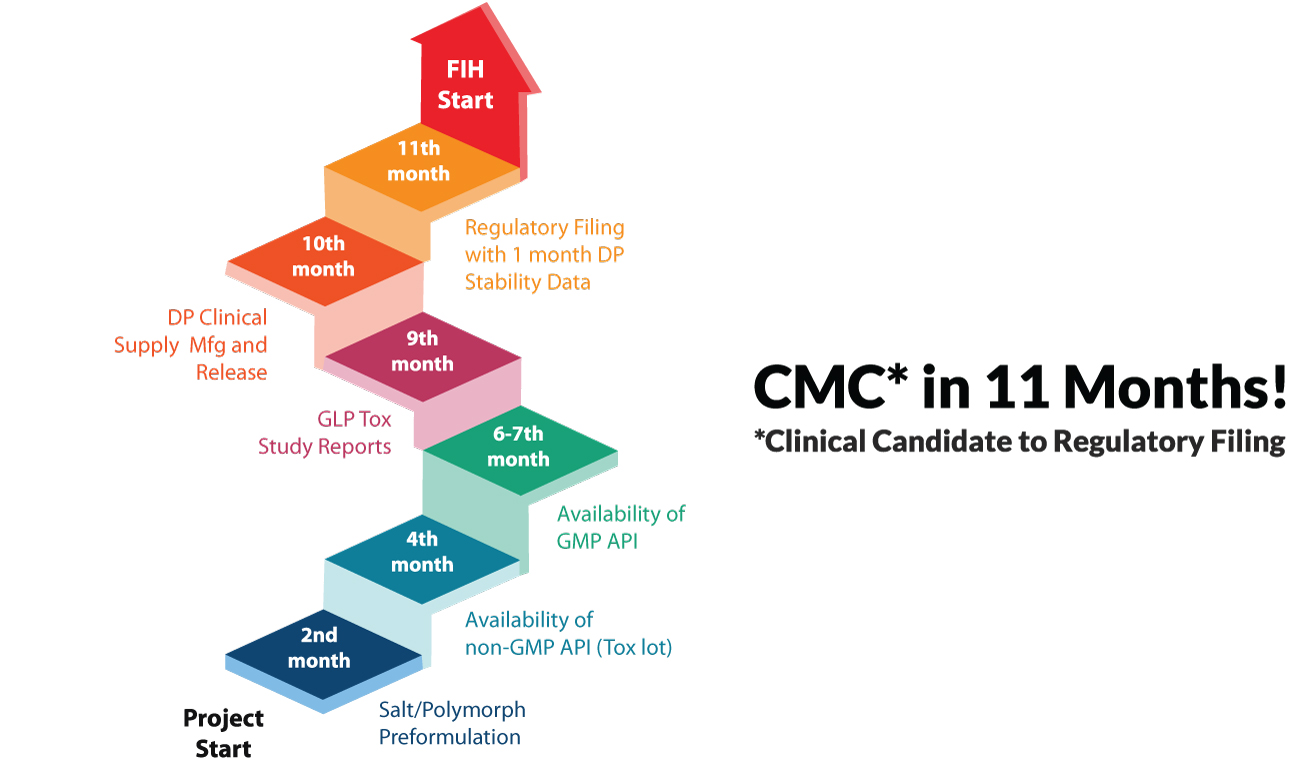
Integrated CMC development for a US pharma major – Successful IND filing & FIH study in 11 months
The lead compound under consideration was a highly soluble and permeable BCS class I molecule. The program objective was IND filing, with a formulation strategy focused on uniformity and stability. The oral route of administration was preferred as the target was seen to be dose proportional after oral dosing in preclinical animal models.
Pre-formulation profiling and target assessment added significant value in the drug substance development phase by uncovering insights that enabled yield improvement of 10% at two intermediate steps. Heat cycles were established to achieve a Chiral purity >98.0% (at the n-1th step). A deep understanding of the target properties helped identify that excess addition of a specific reagent facilitated reaction completion. The integrated CMC program design enabled validation of 5 analytical methods to be completed in a 11-month timeframe. The desired form was successfully achieved in the final crystallization, with the use of wet milling process to obtain the desired particle size (d90 < 50 μm) with no form change.
A dry granulation process was deployed in the drug development to achieve good flow and compressibility and a geometric dilution approach helped in achieving content uniformity. A common blend was used for 1 mg and 5 mg strengths (1% drug load) and a separate blend for 0.3 mg strength (0.3% drug load). A content uniformity value of 5.3 was achieved for 0.3 mg strength. Excipient selection based on chemical compatibility study was a key value driver at this stage.
Identical analytical methods were used for both DS and DP for monitoring degradation. DS and DP were found to be stable for 6M at 40/75 and 2 years at 25/60. No alkaline impurities were detected in tablet form with citric acid as a pH stabilizer. IND was successfully filed and FIH studies initiated just 11 months after the start of the program.
CMC development for small virtual biotech from Europe – IMPD filing in 12 months
Pre-formulation profiling of the drug molecule revealed poor aqueous solubility. Hence solubility enhancement was one of the key development objectives. The solution formulation was thus designed for enhanced bioavailability for toxicity studies. The formulation for GLP toxicity and FIH was to be the same. A Drug-in-bottle formulation was to be used for Phase I (for onsite compounding in the clinic), with a target concentration of 20 mg/ml. Short term stability of formulation was determined to be enough for toxicity dosing, with long term stability of powdered drug-in-bottle formulation targeted for IMPD regulatory filing.
Clear development objectives and regulatory guidelines being set in place helped in the optimization of the process. Some of the key enhancement included increasing selectivity of substitution from 30% to 75% by selecting the right reaction base with respect to process parameter, introducing Suzuki coupling reaction in n-3rd step to control palladium instead of in the final step, and increasing Selectivity of Grignard reaction from 70 to 95% by introducing LiCl as base for the reaction.
98.5%+ purity was achieved for all development and GMP batches (purity of tox was deliberately achieved at 97.5% to qualify higher levels of impurities). A stable dihydrate form was obtained since the anhydrous form had significantly lower yield. Tox lot was found to be physically and chemically stable for 6 months at accelerated conditions. The cGMP campaign was successfully manufactured with desired quality attributes as laid out at the target assessment stage.
The co-solvent approach was established for Drug in Bottle (DIB) formulation. Drug-vehicle-container closure compatibility and formulation stability at 25°C/60%RH and 40°C/75%RH conditions were established with in-use stability of up to 24hrs. Both dihydrate and anhydrous forms were found to be soluble in the vehicle (20 mg/ml) but the dihydrate form was selected based on solubility, stability, and ease of manufacturing. In-vitro taste sensing of the formulation was also conducted and it was found to be bitter. A bitrex solution was used as the clinical dosing strategy.
The entire CMC development including tox and drug substance & drug product clinical campaigns was completed in 12 months, and the IMPD regulatory compilation was successfully drafted and shared with the client.
Summary
As demonstrated through the 2 case studies, an integrated CMC strategy can uncover early insights that will help avoid challenges in early phase development and expedite the time to filing. Critical factors to consider when picking your integrated CMC partner include a track record of successful IND/IMPD filings, the right team with experienced scientists and leadership, and integrated workflow with a dedicated project management team. Thus, partnering with a one-stop-shop CRO partner such as Syngene for end-to-end CMC development services (DS, DP, Analytical, DMPK, GLP tox, GMP clinical Supplies Manufacturing, Stability, Regulatory and QP) can exponentially increase the chances of success of your small molecule development program.